UnoSeqExpression profiling with next generation sequencing without a reference genome |
Site Map
Publication
- Into the unknown: Expression profiling without genome sequence information in CHO by next generation sequencing, Fabian Birzele, Jochen Schaub, Werner Rust, Christoph Clemens, Patrick Baum, Hitto Kaufmann, Andreas Weith, Torsten Schulz and Tobias Hildebrandt, Nucleic Acids Research, 2010
Author
- Fabian Birzele
- gene_expression_all.out: File containing expression values summarized for every gene of the reference organism in the a tab separated format: GeneID RPKM #ReadsMappingToGene RPKMForUniqueReadsOnly #ReadsMappingUniquely #ReadsMappingMultipleTimes. The file may be used to analyze gene expression and differentially expressed genes.
- sorted_reads_for_genes.out: File containing reads sorted to every reference organism gene (file format as shown above) which can be used for knowledge based assembly.
- Perform a de-novo assembly of your reads, all lanes together, using the ReadAssembly tool
- Annotate de-novo contigs using BLAST and parse data using the BLAST parser
- Perform a first mapping round using Bowtie
- Perform a first read analysis step (using ReadAnalysis) and employing your reference organisms (at least one well annotated genome like mouse, human, rat and some other, less well annotated but closer related species) and the de-novo assembly
- Perform a knowledge-based assembly using the ReadAssembly tool, uniquify contigs from de-novo and knowledge-based in a final assembly
- Perform a second mapping round of your reads against the final transcriptome assembly using Bowtie
- Perform a second read analysis step using the ReadAnalysis pipeline and employing your final transcriptome assembly.
- examples/project_data: contains example files for configuration and orthologous mappings of an organism or a contig set onto the reference genome. In that case Rat and a CHOAssembly are mapped onto the respective Mouse genes. The config file defines a run where Mouse is used as reference organism and two additional sources, i.e. an assembly and Rat are used as further references.
- examples/lane: this folder contains example data for one single gene and reads mapping to that gene in Mouse, Rat and CHO (*.bowtie files). Additionally all read sequences which are mapped to that gene are contained in a file called lane.fasta. Those are examples for files which need to be present for every lane you want to process with UnoSeq given the corresponding configuration file (where i.e. the three reference sets are specified).
0. Content of this manual
1. Requirements: describes the requirements of UnoSeq and its dependencies on third-party tools
2. General Introduction: potential use cases of UnoSeq
3. Read Assembly: how to perform different assembly strategies using UnoSeq and Velvet
4. Contig annotation using BLAST: UnoSeq classes for BLAST-based contig annotation
5. Read mapping analysis and Expression profiling: UnoSeq classes to perform expression analysis in organisms without reference genome
6. Suggested Workflow: use case how the different parts of UnoSeq may play together
7. Examples: short walk through the example data provided with the package
8. Remarks: additional remarks
1. Requirements
UNOSEQ is fully implemented in Java and requires at least Java 1.6 to run properly. The software should run on any LINUX / UNIX system where an appropriate Java Runtime Environment is available which can be downloaded from http://java.sun.com.
Several steps also require for large main memory resources, so 10GB are good, 20GB are better, 40GB are surely enough. Even though UNOSEQ is implemented in Java, it relies on several external programs. In more detail those are:
Bowtie: Very well documented and free software to map short reads onto a set of reference sequences. We use this tool to map our reads onto different sets of reference transcriptomes but you may also use any other software to do so. In that case you would have to change the code parsing read mapping information as described below. The software can be downloaded from http://bowtie-bio.sourceforge.net/. In our experiments we used the 0.11 release but higher versions should work as well.
Velvet: Very good software to perform assembly of short reads even for millions of short read sequences. The software can be downloaded at http://www.ebi.ac.uk/~zerbino/velvet/
BLAST: We use BLAST to annotate contigs obtained in the read assembly steps to a set of reference sequences (if possible from a well annotated organism like Human or Mouse). Download at: http://blast.ncbi.nlm.nih.gov/Blast.cgi?CMD=Web&PAGE_TYPE=BlastDocs&DOC_TYPE=Download
For instructions how to install the different tools we would like to point you to the specific documentation of the corresponding software.
2. General Introduction - What can UNOSEQ do for you?
UNOSEQ is a library of java classes which can help you to analyze mRNAseq data generated for organisms for which no reference genome is available. Therefore, the package may contain useful classes to perform expression profiling and transcriptome analyses in ANY ORGANISM, as long as you have genomes and / or transcriptomes of (closely) related organisms (the closer the better) available.
As the genome of interest is not known, the focus of this library is expression profiling of genes and transcriptome assembly only, ignoring the analysis of splice variants, ncRNAs and so on. We have used the library to profile gene expression in the Chinese hamster, which is described in the publication below, where you will also find more details on the workflow and the rationale behind all the different steps.
Into the unknown: Expression profiling without genome sequence information in CHO by next generation sequencing, Fabian Birzele, Jochen Schaub, Werner Rust, Christoph Clemens, Patrick Baum, Hitto Kaufmann, Andreas Weith, Thorsten W. Schulz, Tobias Hildebrandt, Nucleic Acids Research, 2010
Parts of the software can of course also be used to analyse standard mRNAseq data and to do expression profiling there but that is not the focus and better suited packages may be available.
In the case of our CHO transcriptome project, we used the following workflow:
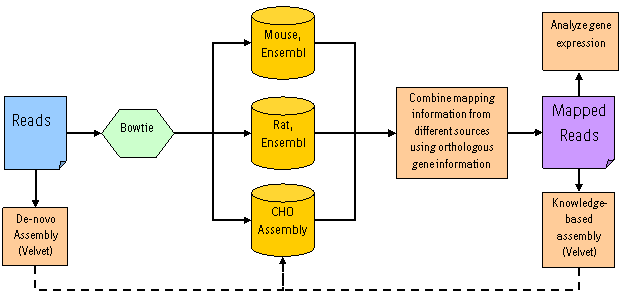
As shown above, we propose to use reference organisms (in that case Mouse and Rat) in combination with transcriptome information generated through assembling short read data in two different ways (De-novo and knowledge-based as described below) to finally profilie gene expression in the corresponding organism. Therefore, our analysis pipeline consists of two steps: read assembly (de-novo and knowledge-based) and read mapping to reference transcriptomes and assembly sequences.
The UNOSEQ library provides Java classes to perform all the different steps and to integrate information from the single parts. In the following we will shortly outline how the different parts of the pipeline can be used and which classes can easily be modified to fit your needs.
3. Read assembly
In order to provide an easy access to the assembly capabilities of Velvet we have written a class called ReadAssembly in the unoseq package. This class provides three funtionalities throught the command line. All functionalities are finally provided by running Velvet, so a running Velvet installation must be available on your system.
In order to assemble a file of FASTA sequences in a de-novo fassion you can use the following program call (of course you have to provide the appropriate parameters):
java -cp . -Xmx10000m unoseq.ReadAssembly -assembleFasta -f input file (FASTA) -o output file for contigs generated -t temporary directory where Velvet will write files to (will NOT be deleted automatically!) -v path to your Velvet installation
A knowledge-based assembly, in contrast to de-novo assembly, is performed not on all reads generated in an experiment but is in our definition limited to all reads assigned to a specific gene. The mapping pipeline (see below) will produce a file (sorted_reads_for_genes.out) where reads are sorted w.r.t. the genes they are annotated to. An example for the file format is shown below, where >GeneID indicates a new gene and all reads assigned to that gene follow afterwards, one read per line.
>ENSMUSG00000049932
CGGTGAATCCCTATCTGGACTGAGCNTCTACTGGCTTCTGA
AAACTCCCCACTACCTAAGGTTCTAAACTTGGGATAGATGC
TAGGGATTCACCGACTTGTGCTGGTATCTAGGTGTTTGGGT
Given such a file, one can use the ReadAssembly class to perform a knowledge-based assembly for all reads annotated to a gene in various files. The method is called something like:
java -cp . -Xmx10000m unoseq.ReadAssembly -knowledgeBasedAssembly -g list of genes -o contig output file -t temporary directory where Velvet will write files to (will NOT be deleted automatically!) �-v path to your Velvet installation -r space separated list of files containing sorted read data for genes
Having performed a set of de-novo and / or knowledge-based assemblies one usually comes up with a set of contigs assigned to a specific gene. Those contigs may overlap and contain redundant information. In order to uniquify a set of contigs the ReadAssembly class provides a final command line option:
java -cp . -Xmx10000m unoseq.ReadAssembly -uniquifyContigs -c ContigInputFile -o ContigOutputFile
Please note that all the assembly functionalities are based on using Velvet as assembly program. If you want to use another assembly tool or if you want to perform your assembly using different parameters you need to modify the specific methods and arguments in the ReadAssembly.java code.
4. Contig Annotation using BLAST
To annotate contigs obtained in a de-novo assembly to a set of well characterized genes we use BLAST, in our case against Mouse transcriptome information (Ensembl+RefSeq). As Hamster and Mouse are related but with some evolutionary distance we used BLAST parameters adopted for more distant sequence searches: blastall -p blastn -d blast database -i contig file -m 7 -o output file -b 5 -e 10e-7 -W 16 -r 2 -q -3 -G 5 -E 2
You can perform your sequence search with whatever parameters you find to fit to your needs. The only two things which are important (if you want to use that part of the UNOSEQ package) is that the output is in XML format (option -m 7) and that the ids of the reference sequences in your db contain the gene name at the first position, separated by a | from any other information.
In that case you can use the unoseq.parser.BLASTAssemblyMappingParser class to parse the BLAST output and annotate your set of initial contigs. The command line call looks like this:
java -cp . -Xmx1000m biokit.ngs.pipeline.parser.BLASTAssemblyMappingParser BLASTOutputXMLFile OriginalFileWithContigs.fasta AnnotatedContigsOutput.out NonAnnotatedContigsOutput.out
The result of that call will be two files containing annotated contigs, namely those which have found a good, possibly unique match partner in your reference sequence set. All other contigs which map to too many other reference sequences (are not specific enough), or which do not find a similar sequence at all are written to the non-annotated contig file.
Please note that you don't have to perform that step using the class provided here but any other way of annotating contigs to reference sequences will be ok.
5. Read mapping analysis and expression profiling
In order to do expression profiling in your organism with an unknown genome and transcriptome, you should try to gather as many sequences either originating from this organism i.e. by assembling reads as outlined above and should integrate this information with transcriptome sequences from closely related organisms.
We perform those mappings of reads against reference sequence sets using Bowtie (see above) and usually use transcript sequences only for such mappings. In order to integrate an arbitrary set of transcriptomes of sequenced organisms and contig sets (de-novo and / or knowledge-based) to a final expression analysis and profiling of the organism you have data for we have written the unoseq.UnoseqReadAnalysis class.
This class requires for the following data regardless which tools you have used beforehand.
a) Configuration File
The configuration file is a simple text file which contains the following parameters (# indicates a comment!)
# organism definitions
# name|geneNamePrefix|referenceOrganism|requiredQuality|orthologMappingFile;nextName|;
organisms=Mouse|ENSMUSG|true|0.9|xy;Rat|ENSRNOG|false|0.95|orthologsWithMouseFile;
# contig set definitions
# contigSetName|contigSetType|contigToGeneMappingFile|requiredQuality;nextContigSet|...
contigSets=CHO assembly|Assembly|contigIDToGeneMappingFile|0.95;...
# read assembly
temporaryDirectory=/tmp /
velvetPath=/myvelvetinstallation/
b) Mapping files
For every organism and every contig set specified above, you have to provide a corresponding mapping file of the reads in your dataset onto the genes / transcripts of the corresponding organism. Those files must be placed all in the same directory and must satisfy certain naming conventions. They must be called like the organism or contig set names specified in the config file followed by ".transcriptome.bowtie" in the case of organisms and by .contigset.bowtie in the case of contig sets. For example the mapping to mouse should be available as a file which is called Mouse.transcriptome.bowtie. Or the file with the matches to the CHO assembly should be available in a file called CHOassembly.contigset.bowtie.
The files need to be standard Bowtie output files which look like this:
1_1_4_1259_1 - ENSMUSG00000037815|18|ENSMUST00000042345|18 2210 TGT... III... 0
The only important thing is the id of the reference sequence (which must correspond to the gene name) and some other information. The gene name must be the first token separated by |. In the example, the gene name is ENSMUSG00000037815.
Read quality information is parsed as well and is used to filter reads which do not suffice a certain quality criterion.
c) Ortholog mapping
In order to combine mapping information from different organisms the user has to specify for every organism except for the reference organism and for every contig set a file which defines the mapping of genes and contigs to reference organism genes. For organisms, those files may contain a 1 to n mapping in the following file format (one line per gene):
OrganismGeneID ReferenceOrganismGeneIDOne|ReferenceOrganismGeneIDTwo
For contigs this mapping must be unique and is specified as shown below (one line per contig)
ContigID ReferenceOrganismGeneID
d) Reference gene lengths
During expression profiling, we compute RPKM values as defined by Mortazavi et al. To compute those values the length of a gene / transcript is important. Therefore, you have to provide a mapping of reference organism gene to the respective exonic length of the gene, or the length of the longest transcript. This is done by a simple file which has the following format:
ReferenceOrganismGeneID GeneLengthInBases
Perform a read analysis
If all the required files and data has been prepared, you can run a read analysis using the following command line call:
java -cp . -Xmx8000m unoseq.ReadAnalysis -mRNASeq-workflow-unknown -o output directory also containing mapping files -c config file as specified above -r read sequences (FASTA file) -l file containing reference gene to length mapping [-i list with interesting gene ids (one per line) others will be ignored]
This analysis will run for a while. You have to make sure that you provide enough main memory (extend with -Xmx30000m if necessary). The results are written to the specified output directory. Several output files are generated during the run, the two most important ones are:
All other files might carry interesting information but are not important for any futher steps.
6. Suggested Workflow
From our experience the follwing workflow has been proven to be successful:
From that point on you can use the read counts and RPKM values for gene expression profiling and all other interesting steps. To identify significantly regulated genes we would recommend the use of SAGEBetaBin (http://www.vision.ime.usp.br/~rvencio/SAGEbetaBin/) or the DEGseq package (http://bioinfo.au.tsinghua.edu.cn/software/degseq/).
7. Example
In the example folder you'll find some example data to get started right away in testing UnoSeq. The following example data is provided:
Given that data you can simply run the analysis pipeline with the following call:
java -cp . unoseq/UnoseqReadAnalysis -mRNASeq-workflow-unknown -o examples/lane/ -c config/ReadAnalysisPipline.config -r examples/lane/lane.fasta -l examples/project_data/mouse_gene_lengths.out
In the output directory examples/lane you'll find all the data written by the pipeline in the course of the analysis, including the gene_expression_all.out file where your gene expression information can be found.
8. Remarks
If you have any questions regarding the software please contact me at fabian dot birzele at boehringer-ingelheim dot com or through sourceforge.
If you find the software useful for your own research, please cite
Into the unknown: Expression profiling without genome sequence information in CHO by next generation sequencing, Fabian Birzele, Jochen Schaub, Werner Rust, Christoph Clemens, Patrick Baum, Hitto Kaufmann, Andreas Weith, Thorsten W. Schulz, Tobias Hildebrandt, Nucleic Acids Research, 2010